
L’Epidemia che Definisce la Nostra Epoca
Tratto da un documento pubblicato sulla biblioteca Zenodo.org scaricalo qui in Inglese
ACQUISTA E SCARICA LA TRADUZIONE IN ITALIANO CLICCA QUI
Adiuvanti di Alluminio, Autoimmunità e Disturbo dello Spettro Autistico: Un’Analisi Completa dei Meccanismi, della Neuropatologia e degli Aspetti Legali
Autori: Brian Hooker PhD*, James Neuenschwander MD, Karl Jablonowski PhD, Martha Herbert MD PhD, Heather Ray, Martin Roberts, Clayton Baker MD, Christopher Shaw PhD
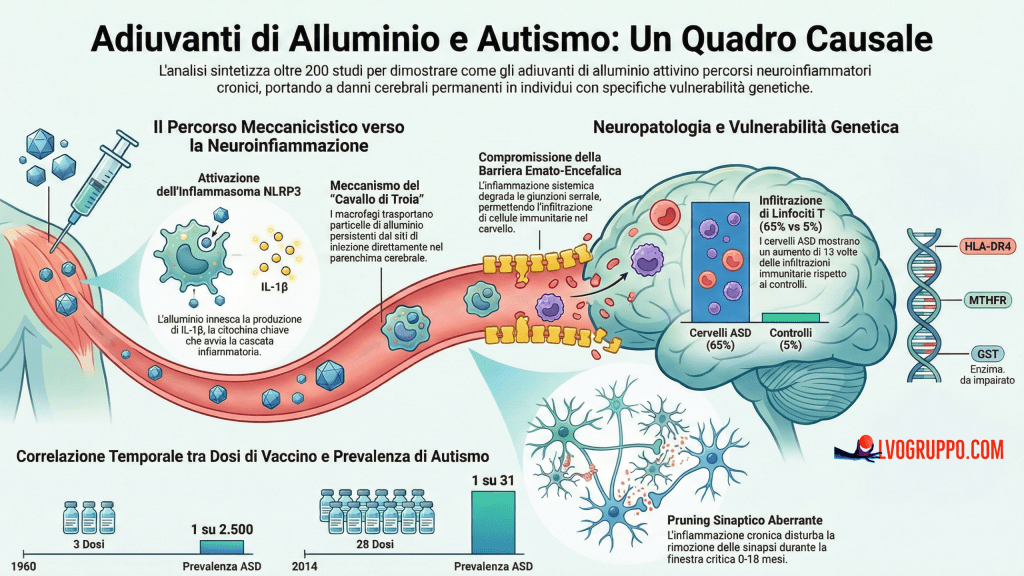
Nel 1960, il disturbo dello spettro autistico (ASD) rappresentava una condizione clinica rara, con una prevalenza stimata tra i 2 e i 4 casi ogni 10.000 bambini. Fast forward al 2025: i dati di sorveglianza del Centers for Disease Control and Prevention (CDC) statunitense descrivono una realtà radicalmente diversa. Per i bambini nati nel 2014, la diagnosi di autismo viene posta con una frequenza di 1 caso ogni 31 individui, pari al 3,2% della popolazione pediatrica. Questo incremento di 80 volte in poco più di mezzo secolo rappresenta uno dei più drammatici cambiamenti epidemiologici nella storia della medicina moderna, un fenomeno che non può essere liquidato come semplice risultato di criteri diagnostici più inclusivi o di una maggiore consapevolezza clinica.
La comunità scientifica si trova di fronte a un’emergenza di salute pubblica che impone una domanda fondamentale e scomoda: quale fattore ambientale abbiamo sistematicamente trascurato? La ricerca neuropatologica e immunologica d’avanguardia sta convergendo su un sospettato specifico e controverso: gli adiuvanti di alluminio. Utilizzati per decenni per potenziare la risposta immunitaria nei vaccini, questi composti sono oggi al centro di una mappatura meccanicistica che rivela come, in soggetti geneticamente vulnerabili, possano innescare una cascata neuroinfiammatoria cronica capace di deragliare il delicato programma di sviluppo del cervello infantile.
Questo articolo si propone di analizzare in modo approfondito e critico le evidenze scientifiche disponibili, esaminando i meccanismi biologici, le implicazioni cliniche, i gap normativi e le responsabilità istituzionali che emergono da questa complessa questione.
L’obiettivo non è quello di fornire risposte definitive su un tema ancora oggetto di dibattito scientifico, ma piuttosto di illuminare le zone d’ombra, sollevare le questioni che meritano maggiore attenzione e stimolare una riflessione informata su un tema che tocca la vita di milioni di famiglie in tutto il mondo.
La Correlazione Statistica: Più di una Semplice Coincidenza
L’espansione del calendario vaccinale pediatrico ha proceduto con una sincronia inquietante rispetto all’impennata dei casi di disturbo dello spettro autistico. Nel 1960, il piano vaccinale prevedeva soltanto 3 dosi entro i 18 mesi di età, somministrate attraverso il vaccino DTP (difterite-tetano-pertosse). Oggi, secondo le raccomandazioni aggiornate del CDC, i bambini ricevono 28 dosi entro i primi due anni di vita, distribuite tra 12 diversi prodotti vaccinali che includono epatite B, DTaP, Haemophilus influenzae tipo b, pneumococco, morbillo-parotite-rosolia, varicella e vaccini antinfluenzali.
La progressione temporale di questa espansione vaccinale è supportata da un dato statistico che, in qualsiasi altro campo della medicina ambientale o tossicologica, susciterebbe un allarme immediato e una risposta regolatoria urgente. L’analisi di regressione e correlazione dei dati di sorveglianza CDC sui trend di prevalenza dell’ASD e sui tassi di vaccinazione a livello statale negli Stati Uniti rivela un coefficiente di correlazione di Pearson pari a r = 0,91, con un valore di significatività statistica p = 0,0015. In termini epidemiologici, un valore di correlazione di 0,91 indica un’associazione positiva quasi perfetta tra le due variabili.
È fondamentale sottolineare che la correlazione statistica, per quanto robusta, non costituisce di per sé prova di causalità. Tuttavia, la solidità di questo legame numerico, unita all’assenza di qualsiasi studio prospettico di coorte che abbia mai esaminato specificamente gli effetti neuroevolutivi del carico cumulativo di adiuvanti di alluminio, evidenzia una lacuna normativa e scientifica inaccettabile. Gli attuali limiti di sicurezza per l’alluminio nei prodotti biologici non derivano da studi moderni di neurotossicità dello sviluppo, ma sono stati ereditati attraverso il principio del “grandfathering” da dati sulla immunogenicità risalenti agli anni ’60 e ’70, un’epoca che ignorava totalmente la biologia dell’inflammasoma NLRP3, la dinamica della barriera emato-encefalica e i meccanismi di traslocazione macrofagica.
L’analisi dei potenziali fattori confondenti rivela che questi non sono sufficienti a spiegare l’80 volte incremento osservato. Le modifiche ai criteri diagnostici del Manuale Diagnostico e Statistico dei Disturbi Mentali (DSM), dalla terza alla quinta edizione, sono avvenute in modo discreto e non esponenziale, non accounting per l’aumento continuo documentato nell’intero periodo 1960-2025. La cosiddetta “sostituzione diagnostica”, ovvero la redistribuzione di bambini precedentemente diagnosticati come “disabili intellettuali” nella categoria dell’autismo, spiega al massimo un declino del 20-30% della prevalenza di disabilità intellettiva, numeri insufficienti per giustificare un incremento dell’8000% dell’ASD. Fattori ambientali alternativi come pesticidi, inquinamento atmosferico e sostanze chimiche endocrine-disruptive non sono stati sistematicamente introdotti in modo sincrono con l’espansione del calendario vaccinale nel timeframe considerato.
ACQUISTA E SCARICA LE SLIDE COMPLETE QUI
Il Paradosso Regolatorio: Protezione dei Produttori e Assenza di Test di Neurotossicità
Il quadro normativo che governa la sicurezza dei vaccini pediatrici negli Stati Uniti presenta un paradosso strutturale che merita un’attenta analisi critica. Il National Childhood Vaccine Injury Act (NCVIA) del 1986, codificato nel Title 42 degli United States Codes alle sezioni 300aa-1 e successive, ha istituito un programma di compensazione no-fault in cambio dell’immunità legale dei produttori vaccinali. Questo accordo quid pro quo ha di fatto creato un sistema in cui i produttori non affrontano responsabilità civile per danni da vaccino, trasferendo l’onere finanziario al governo federale attraverso il Vaccine Injury Compensation Trust Fund.
La giurisprudenza consolidata, culminata nella sentenza della Corte Suprema Bruesewitz v. Wyeth del 2011, ha ulteriormente rafforzato questa immunità stabilendo che le pretese basate sul difetto di progettazione (design defect claims) sono precluse anche quando esistono alternative progettuali più sicure tecnicamente fattibili. Questa architettura legale ha creato quello che gli esperti definiscono un “regulatory double-bind”: da un lato, le agenzie regolatorie come la Food and Drug Administration (FDA) e il CDC non possiedono l’autorità statutaria per richiedere test di neurotossicità dedicati per componenti vaccinali “grandfathered” come gli adiuvanti di alluminio; dall’altro, i produttori non hanno alcun incentivo economico a investire nella sicurezza ottimale dei propri prodotti, essendo protetti da qualsiasi responsabilità per danni.
La regolazione FDA per i vaccini, codificata nel 21 CFR § 610.15(a), richiede genericamente che gli adiuvanti “non influenzino negativamente la sicurezza o la potenza” del prodotto. Questo criterio ambiguo viene valutato attraverso trial clinici a breve termine focalizzati su reazioni acute quali febbre, eritemi e irritabilità, trascurando sistematicamente gli esiti neuroimmunologici cronici che potrebbero manifestarsi mesi o anni dopo l’esposizione. Il principio del “grandfathering” esenta i sali di alluminio dai moderni standard tossicologici basandosi esclusivamente sul loro uso storico, una logica che appare gravemente inadeguata considerando che la popolazione pediatrica odierna è esposta a un carico cumulativo di alluminio che varia tra 2.505 e 5.175 microgrammi entro i primi 18 mesi di vita, una finestra critica di vulnerabilità neurologica mai considerata quando questi limiti furono originariamente stabiliti.
Il confronto con gli standard applicati ad altre sostanze chimiche e farmaci rivela un divario normativo impressionante. Le linee guida dell’Agenzia per la Protezione Ambientale (EPA) e dell’Organizzazione per la Cooperazione e lo Sviluppo Economico (OECD) richiedono studi di neurotossicità evolutiva per sostanze che: producano effetti neurotossici in studi di tossicità a dosi ripetute; possiedano un target farmacologico nel sistema nervoso centrale; mostrino alterazioni neuropatologiche o comportamentali in studi preclinici; o dimostrino accumulo cerebrale o effetti sui sistemi neurotrasmettitoriali. L’alluminio soddisfa tutti e quattro questi criteri secondo le evidenze scientifiche disponibili, eppure rimane esente da qualsiasi obbligo di test di neurotossicità evolutiva nel contesto vaccinale.
Il “Cavallo di Troia”: Come l’Alluminio Viaggia dal Muscolo al Cervello
Per decenni, il paradigma scientifico dominante si è basato su un errore tossicocinetico fondamentale che ha plasmato la percezione della sicurezza degli adiuvanti di alluminio. L’ipotesi del “deposito” (depot effect), proposta inizialmente per spiegare il meccanismo d’azione degli adiuvanti, postulava che le particelle di alluminio rimanessero localizzate nel sito di iniezione, creando un serbatoio antigienico prolungato da cui l’antigene veniva gradualmente rilasciato per stimolare il sistema immunitario. Questa concezione implicava che l’alluminio fosse essenzialmente innocuo perché confinato nel tessuto periferico.
La realtà biomedica si è rivelata molto più complessa e preoccupante. L’alluminio utilizzato come adiuvante nei vaccini non è in forma solubile, ma particolata, una configurazione che conferisce resistenza alla clearance renale e favorisce la biopersistenza tissutale. Il meccanismo attraverso cui queste particelle raggiungono il sistema nervoso centrale è stato descritto elegantemente dal gruppo di ricerca di Romain Gherardi presso l’Università Paris-Sud e può essere assimilato a un “Cavallo di Troia” biologico.
Il processo inizia con l’iniezione intramuscolare del vaccino contenente particelle di alluminio. Nel tessuto muscolare, i macrofagi identificano le particelle come Damage-Associated Molecular Patterns (DAMPs) e le fagocitano attraverso un processo di endocitosi. A differenza di quanto previsto dai modelli tradizionali, i macrofagi non neutralizzano né eliminano il metallo; al contrario, lo trasportano attraverso il sistema linfatico verso i linfonodi drenanti, e successivamente nel circolo sanguigno. Sotto la spinta della chemochina CCL2 (Chemokine C-C Motif Ligand 2), i macrofagi carichi di alluminio migrano verso il sistema nervoso centrale, attraversando la barriera emato-encefalica (BEE) e infiltrandosi nel parenchima cerebrale.
Studi sperimentali condotti su modelli animali hanno documentato che le particelle di alluminio raggiungono il cervello entro 21 giorni dalla somministrazione intramuscolare e possono persistere nel parenchima cerebrale per periodi superiori a un anno. Una volta arrivate nel tessuto cerebrale, queste particelle non restano inerti ma continuano a stimolare una risposta infiammatoria cronica, attivando le cellule immunitarie residenti e perpetuando un ciclo di neuroinfiammazione che mina le fondamenta dei modelli tossicologici tradizionali basati sulla clearance rapida dell’alluminio solubile.
Un fattore sinergico critico in questo processo è rappresentato dal polisorbato 80 (noto anche come Tween 80), un eccipiente utilizzato in numerosi preparati vaccinali come emulsionante e stabilizzante. Questo tensioattivo è in grado di modulare la permeabilità della barriera emato-encefalica attraverso il meccanismo del “nanoparticle overcoating”, facilitando l’attraversamento delle particelle rivestite verso il sistema nervoso centrale. Sebbene le quantità di polisorbato 80 nei vaccini siano considerate “tracce” dai regolatori, la sua azione sinergica con l’alluminio nel contesto del meccanismo del Cavallo di Troia merita un’attenta rivalutazione tossicologica.
L’Interruttore dell’Infiammazione: L’Inflammasoma NLRP3 e la Cascata Citochinica
A livello molecolare, l’alluminio agisce come un potente attivatore dell’immunità innata attraverso l’attivazione dell’inflammasoma NLRP3 (NOD-like Receptor Family Pyrin Domain Containing 3), un complesso multiproteico che funge da sensore di pericolo intracellulare. Questo meccanismo rappresenta il cuore della risposta infiammatoria sistemica indotta dagli adiuvanti e costituisce il legame fondamentale tra l’esposizione periferica e la patologia cerebrale.
L’inflammasoma NLRP3 è composto da tre componenti principali: la proteina sensore NLRP3, che rileva i segnali di pericolo; la proteina adattatrice ASC (Apoptosis-associated Speck-like Protein containing a CARD), che media l’olimerizzazione del complesso; e la pro-caspasi-1, l’effettore enzimatico che viene attivato mediante cleavage. L’assemblaggio di questo complesso multiproteico avviene in risposta a segnali di pericolo di natura diversa, e l’alluminio è in grado di attivarlo attraverso tre percorsi convergenti.
Il primo percorso è quello fagocitario. Quando i macrofagi fagocitano le particelle di alluminio, la natura cristallina e particolata del metallo induce la destabilizzazione della membrana lisosomiale, causando la rottura del lisosoma e il rilascio nel citoplasma della catepsina B, una proteasi lisosomiale che funge da potente segnale di attivazione per NLRP3. Il secondo percorso coinvolge lo stress ossidativo mitocondriale: l’alluminio interferisce con la funzione mitocondriale, causando la disfunzione della catena di trasporto degli elettroni e la generazione massiccia di specie reattive dell’ossigeno (ROS), che a loro volta ossidano lipidi e proteine di membrana, creando DAMPs aggiuntivi necessari per l’assemblaggio dell’inflammasoma. Il terzo percorso è quello del flusso ionico: l’alluminio altera la permeabilità delle membrane cellulari, causando un efflusso di potassio che rappresenta il segnale canonico per l’attivazione di NLRP3, inducendo i cambiamenti conformazionali necessari per l’olimerizzazione del complesso.
Una volta attivato, l’inflammasoma NLRP3 scatena l’attivazione della caspasi-1, che a sua volta cliva la pro-IL-1β e la pro-IL-18 nelle loro forme mature e bioattive. L’interleuchina-1 beta (IL-1β) merita particolare attenzione come “citochina master” che orchestra la transizione dall’infiammazione periferica alla patologia cerebrale. Gli studi di meta-analisi su individui con ASD hanno rivelato che i livelli sierici di IL-1β sono elevati di 1,56 volte rispetto ai controlli neurotipici, un dato statisticamente significativo che conferma lo stato di attivazione immunitaria sistemica caratteristico di questa popolazione.
L’aspetto più critico identificato dalla neuropatologia moderna è la creazione di un ciclo di feedback positivo autorigenerante. Come dimostrato da ricerche recenti, l’attivazione di NLRP3 e la conseguente disfunzione mitocondriale formano un loop positivo in cui l’infiammasoma attivato frammenta i mitocondri, i quali rilasciano DAMPs mitocondriali (DNA mitocondriale, cardiolipina, citocromo c) che riattivano ulteriormente NLRP3. Questo meccanismo spiega a livello molecolare la natura cronica e non-risolvente della neuroinfiammazione caratteristica della neuropatologia ASD.
Oltre all’IL-1β, la cascata infiammatoria coinvolge altre citochine pro-infiammatorie cruciali che amplificano e sostengono il danno neurologico. L’interleuchina-6 (IL-6) risulta primariamente elevata nel sistema nervoso centrale rispetto alla periferia, fungendo da amplificatore secondario del danno e della disfunzione sinaptica. Il fattore di necrosi tumorale alfa (TNF-α) media la morte cellulare programmata e contribuisce alla disregolazione della barriera emato-encefalica. L’interferone gamma (IFN-γ) amplifica la risposta immunitaria cellulo-mediata e promuove l’infiltrazione di linfociti nel tessuto cerebrale. Questa rete citochinica opera in modo sinergico, creando uno stato infiammatorio cronico che compromette l’omeostasi del sistema nervoso centrale.
La Barriera Emato-Encefalica: Il Cancello che si Apre
L’interleuchina-1 beta svolge un ruolo cruciale nel compromettere l’integrità della barriera emato-encefalica (BEE), una struttura semipermeabile che protegge il cervello da molecole e cellule potenzialmente dannose presenti nel circolo sanguigno. La BEE è costituita da cellule endoteliali cerebrali tenacemente unite da giunzioni serrate (tight junctions) composte da proteine come claudine e occludine, che regolano il passaggio di sostanze dal sangue al cervello.
L’IL-1β induce la downregolazione di queste proteine a giunzione serrata, riducendo drasticamente l’integrità strutturale della barriera e rendendola permeabile a molecole e cellule che normalmente verrebbero bloccate. Parallelamente, l’IL-1β upregola le molecole di adesione vascolare VCAM-1 (Vascular Cell Adhesion Molecule-1) e ICAM-1 (Intercellular Adhesion Molecule-1), che agiscono come ganci molecolari per attrarre e trattenere i leucociti sulla superficie endoteliale. Questo processo facilita la migrazione trans-endoteliale di cellule immunitarie dal sangue verso il parenchima cerebrale.
Un ulteriore meccanismo di compromissione della BEE coinvolge l’interleuchina-17A (IL-17A), prodotta dai linfociti T helper 17 (Th17). Come dimostrato in modelli sperimentali di sclerosi multipla, l’IL-17A danneggia direttamente l’integrità della BEE attraverso l’upregolazione delle metalloproteinasi della matrice (MMPs), enzimi che degradano le proteine della membrana basale e della matrice extracellulare. Questo meccanismo è particolarmente rilevante nel contesto della risposta immunitaria indotta dagli adiuvanti di alluminio, che favoriscono la polarizzazione verso il fenotipo Th17.
La conseguenza pratica di questi meccanismi è l’apertura di un “cancello” che permette ai macrofagi carichi di alluminio, ai linfociti T attivati e alle molecole pro-infiammatorie di penetrare nel sistema nervoso centrale, innescando una cascata di eventi neuropatologici che culminano nel danno cerebrale. Questo processo spiega come un’esposizione apparentemente periferica, come un’iniezione intramuscolare di vaccino, possa tradursi in una patologia del sistema nervoso centrale.
La Neuropatologia dell’Autismo: L’Attacco Immunitario al Cervello
Le evidenze neuropatologiche post-mortem costituiscono il “gold standard” probatorio per comprendere i meccanismi di danno cerebrale nel disturbo dello spettro autistico. Lo studio seminale di DiStasio e colleghi, pubblicato nel 2019, ha documentato un quadro drammatico che supporta l’ipotesi di un attacco immunitario attivo mediato dai linfociti T contro il parenchima cerebrale.
L’analisi neuropatologica ha rivelato una prevalenza di infiltrazione perivascolare di linfociti T pari al 65% nei cervelli di individui con ASD, rispetto a soltanto il 5% nei controlli neurotipici. Questo dato rappresenta un incremento di 13 volte, statisticamente altamente significativo e dotato di una specificità del 95%. Ancora più rilevante, i linfociti T non erano confinati negli spazi perivascolari ma avevano penetrato il parenchima cerebrale, stabilendo contatti diretti con gli astrociti.
L’analisi mediante citometria di flusso e immunoistochimica ha dimostrato un rapporto CD8+/CD4+ elevato (media di 1,8:1 nei soggetti ASD rispetto a 0,8:1 nei controlli), indicando un predominio di linfociti T citotossici CD8+ che sono capaci di uccidere le cellule bersaglio attraverso il rilascio di granzima B e perforina. L’espressione robusta di granzima B, una serinproteasi rilasciata dai linfociti citotossici e richiesta per la lisi cellulare mediata da perforina, fornisce evidenza diretta di un attacco immunitario attivo in corso nel tessuto cerebrale degli individui con ASD.
La microscopia elettronica ha rivelato la presenza di “blebs” astrocitari citotossici nel 32% dei campioni di tessuto ASD esaminati, rispetto a soltanto il 2% dei controlli. Questi blebs rappresentano rotture focali della membrana citoplasmatica degli astrociti, indicando un attacco citotossico diretto a queste cellule gliali cruciali per il funzionamento cerebrale. Gli astrociti non sono semplici cellule di supporto passivo, ma svolgono funzioni attive essenziali per l’omeostasi neuronale: gestiscono oltre il 90% della clearance del glutammato attraverso i trasportatori GLT-1/EAAT2, forniscono supporto metabolico ai neuroni attraverso lo shuttle lattato-astrocita-neurone, e partecipano attivamente alla potatura sinaptica mediata dal complemento.
La perdita di astrociti funzionalmente competenti ha conseguenze devastanti per il cervello in sviluppo. La compromissione della clearance del glutammato innesca una cascata di eccitotossicità, dove l’accumulo eccessivo di questo neurotrasmettitore eccitatorio sovraccarica i recettori NMDA, causando afflusso di calcio mitocondriale, generazione di specie reattive dell’ossigeno e attivazione di proteasi intracellulari che conducono a danno neuronale. Questo meccanismo di eccitotossicità è particolarmente devastante durante le fasi sensibili dello sviluppo, quando i circuiti cerebrali sono in via di formazione e ottimizzazione.
Ulteriori evidenze neuropatologiche includono l’astrogliosi reattiva, documentata dall’aumento dell’espressione della proteina acida fibrillare gliale (GFAP). Studi quantitativi hanno dimostrato un incremento di 1,70 volte dell’mRNA della GFAP nella corteccia prefrontale (p = 0,0049) e di 2,63 volte nel cervelletto (p = 0,0022) dei soggetti ASD rispetto ai controlli. Questa iper-espressione della GFAP è indicativa di uno stato di attivazione astrocitaria cronica, coerente con un processo infiammatorio persistente che caratterizza la neuropatologia dell’autismo.
La Disregolazione della Potatura Sinaptica: Troppe Connessioni, Meno Efficienza
Il sistema del complemento, tradizionalmente noto per il suo ruolo nell’immunità antimicrobica, svolge una funzione parallela fondamentale durante lo sviluppo neurologico: la potatura sinaptica mediata dal complemento (complement-mediated synaptic pruning). Questo processo rappresenta il meccanismo attraverso cui il cervello in sviluppo elimina le sinapsi immature o deboli, raffinando i circuiti neurali e ottimizzando la connettività cerebrale.
Il meccanismo della potatura sinaptica mediata dal complemento opera attraverso una sequenza di eventi finemente regolata. Le sinapsi deboli o immature vengono inizialmente “taggate” mediante deposizione del componente C1q del complemento, che funge da marcatore di riconoscimento. Questo innesca l’attivazione della via classica del complemento, con conseguente clivaggio di C3 in C3b e C3a. Il C3b opsinizza (marca) la sinapsi bersaglio, localizzando ulteriori eventi di amplificazione del complemento. I recettori del complemento (CR1, CR3, CR4) espressi sulla superficie microgliale riconoscono le sinapsi opsonizzate, e i processi microgliali si estendono verso le sinapsi marcate, englobando i terminali presinaptici e le spine dendritiche attraverso la fagocitosi.
Nel contesto dell’autismo, la potatura sinaptica risulta disregolata attraverso meccanismi multipli. Contrariamente ai modelli di eliminazione eccessiva, nell’ASD l’infiammazione cronica e le varianti genetiche inducono un fenomeno di sotto-potatura (under-pruning). La ridotta segnalazione o il malfunzionamento del complesso C1q/C3 impedisce la corretta marcatura delle sinapsi deboli per la fagocitosi microgliale. Varianti nel gene C4, in particolare l’allele nullo C4B che porta a ridotta o assente espressione della proteina C4B, sono state associate a un aumentato rischio di ASD. Queste varianti compromettono l’attivazione efficiente del complemento e il corretto tagging sinaptico, riducendo la capacità di eliminare efficacemente le sinapsi inappropriate durante le finestre di sviluppo critiche.
Il risultato fenotipico di questa sotto-potatura è un eccesso di spine dendritiche immature, che si traduce in iperconnettività locale e ipoconnettività a lungo raggio. L’iperconnettività locale nelle cortecce sensoriali è associata all’ipersensibilità sensoriale caratteristica dell’autismo, mentre l’ipoconnettività a lungo raggio tra regioni cerebrali distanti contribuisce ai deficit di comunicazione sociale e integrazione cognitiva. Studi post-mortem hanno effettivamente documentato un aumento della densità delle spine dendritiche nei cervelli di individui con ASD, coerente con un deficit nella potatura sinaptica durante lo sviluppo.
La neuroinfiammazione indotta dagli adiuvanti di alluminio può contribuire a questa disregolazione attraverso molteplici meccanismi. Lo stato infiammatorio cronico altera l’espressione e la funzione delle proteine del complemento, compromettendo la precisione del processo di tagging. L’attivazione microgliale anomala modifica il comportamento fagocitico di queste cellule, mentre la disregolazione citochinica interferisce con i segnali necessari per l’eliminazione selettiva delle sinapsi. In questo contesto, la finestra critica dello sviluppo (3-36 mesi) assume un’importanza particolare, poiché rappresenta il periodo di massima sinaptogenesi e potatura sinaptica.
La Finestra Critica: Perché i Primi Tre Anni Decidono il Destino
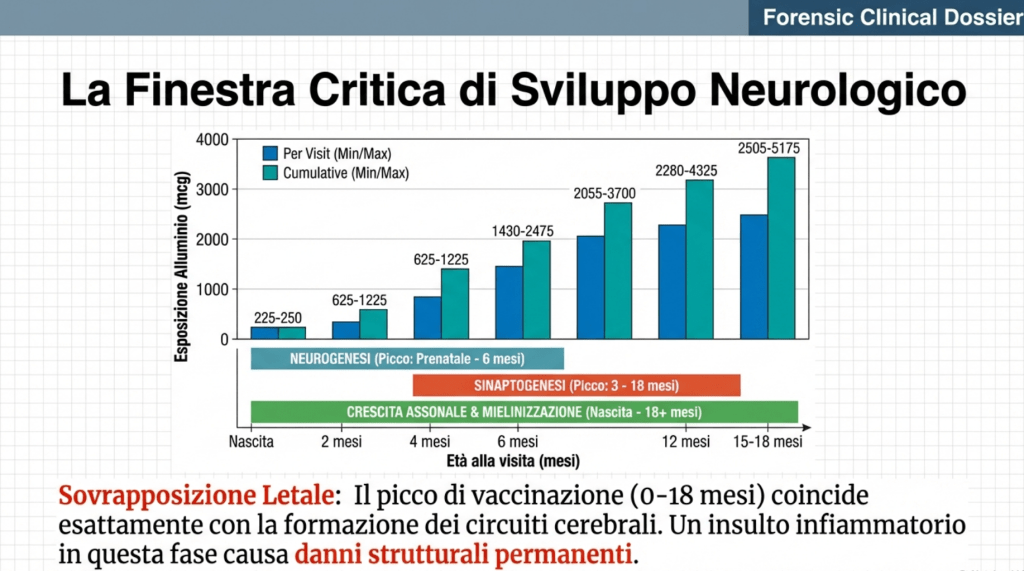
Il cervello umano attraversa una fase di rimodellamento massivo durante i primi anni di vita, caratterizzata da processi di neurogenesi, sinaptogenesi, potatura sinaptica e mielinizzazione che procedono secondo schedule developmental precisi e finemente orchestrati. I lobi frontali e temporali raggiungono il picco della densità sinaptica e dei tassi di potatura tra i 3 mesi e i 3 anni di età, esattamente il periodo in cui il calendario vaccinale pediatrico prevede la concentrazione maggiore di somministrazioni.
La sinaptogenesi, ovvero la formazione di nuove connessioni sinaptiche tra i neuroni, procede a ritmi accelerati durante l’infanzia, con la corteccia visiva che raggiunge il picco sinaptico intorno agli 8 mesi e le aree prefrontali successivamente, verso i 2-3 anni. Parallelamente, la potatura sinaptica elimina approssimativamente il 40% delle sinapsi developmental per stabilire la connettività tipica dell’adulto. La mielinizzazione, il processo di isolamento degli assoni con guaine mieliniche per la conduzione veloce dei segnali nervosi, presenta tassi massimi durante l’infanzia e prosegue durante l’adolescenza.
L’allineamento temporale tra questa vulnerabilità biologica e il picco di esposizione agli adiuvanti di alluminio solleva questioni profonde. Secondo le analisi del carico cumulativo di alluminio derivante dal calendario vaccinale, un bambino che segue le raccomandazioni CDC riceve un’esposizione totale di circa 4,925 mg di alluminio entro i 18 mesi di età. Questo carico si distribuisce in modo non uniforme, con la maggior parte delle dosi concentrate tra la nascita e i 6 mesi, e tra i 12 e i 18 mesi.
I dati mostrano che l’esposizione cumulativa raggiunge livelli di 2.055-3.700 mcg entro i 6 mesi e 2.505-5.175 mcg entro i 15-18 mesi. Questa tempistica coincide precisamente con le finestre critiche per la sinaptogenesi e la potatura sinaptica nelle cortecce associative, nonché con i periodi di rapida mielinizzazione delle vie cerebrali lunghe. La sincronia tra esposizione e vulnerabilità developmental rappresenta un elemento critico per comprendere come l’alluminio adiuvante possa interferire con lo sviluppo neurologico.
Le osservazioni cliniche supportano questa finestra critica. I dati epidemiologici indicano che la maggior parte delle diagnosi di ASD si concentra tra i 14 e i 36 mesi di età, con un picco intorno ai 18-24 mesi. Numerose segnalazioni di genitori documentano una regressione dello sviluppo, ovvero la perdita di competenze precedentemente acquisite come il linguaggio, il contatto visivo e le capacità sociali, in relazione temporale con le vaccinazioni. Uno studio ha rilevato che il 30-40% dei casi di ASD coinvolge una regressione, spesso in prossimità temporale con le vaccinazioni o con altri stressor immunologici.
La Vulnerabilità Genetica: Perché Non Tutti i Bambini Sono Uguali
Se il percorso biochimico dall’esposizione all’alluminio alla neuroinfiammazione è universale, la risposta individuale varia drammaticamente in funzione del profilo genetico. L’autismo è riconosciuto come un disturbo dall’origine poligenica: non esiste un singolo “gene dell’autismo”, ma centinaia di varianti genetiche che influenzano la resilienza dell’individuo. La genetica non è un destino immutabile, ma determina la “soglia di tolleranza” agli insulti esterni, creando popolazioni di individui vulnerabili che pagano il prezzo più alto dell’esposizione ambientale.
L’architettura genetica della suscettibilità si fonda su tre pilastri biologici principali. Il primo riguarda la capacità di detossificazione, ovvero l’efficienza enzimatica nel neutralizzare metalli pesanti e xenobiotici. Il secondo concerne la regolazione della risposta immunitaria, ovvero la precisione nel calibrare l’infiammazione evitando che diventi cronica o autoimmune. Il terzo riguarda le soglie di raffinamento sinaptico, ovvero il controllo molecolare sulla potatura dei circuiti neuronali durante lo sviluppo.
Gli alleli HLA-DR4 rappresentano uno dei fattori di rischio genetico più consistenti. Il sistema HLA (Human Leukocyte Antigen) codifica per le molecole del complesso maggiore di istocompatibilità di classe II, responsabili della presentazione degli antigeni peptidici ai linfociti T helper. L’allele HLA-DRB1*04:01 e i suoi omologhi strutturali conferiscono una capacità amplificata di presentare antigeni ai linfociti CD4+, potenziando le risposte delle cellule T helper. Studi epidemiologici hanno documentato un rischio di ASD aumentato di 2,2-2,9 volte nei portatori di HLA-DR4. Il meccanismo proposto è che questi individui sviluppino risposte T helper più robuste agli antigeni vaccinali; se la vaccinazione avviene nel contesto del mimetismo molecolare o se innesca un epitope spreading, i portatori di HLA-DR4 monterebbero risposte T autoreattive più intense, aumentando la probabilità di infiltrazione linfocitaria e attacco astrocitario.
I polimorfismi del gene MTHFR (Metilentetraidrofolato reduttasi), in particolare le varianti C677T e A1298C, rappresentano un secondo fattore di rischio cruciale, presente nel 40-60% dei casi di ASD. L’enzima MTHFR catalizza la conversione del 5,10-metilentetraidrofolato in 5-metiltetraidrofolato, il substrato per la metionina sintasi che produce metionina dal processo di rimetilazione dell’omocisteina. La variante C677T produce un enzima termolabile con attività ridotta al 35% rispetto al wild-type, mentre la variante A1298C riduce l’attività al 60% in omozigosi. La forma composta 677TT/1298AA riduce l’attività enzimativa in modo analogo all’omozygote 677TT.
Le conseguenze di questi polimorfismi sono molteplici e pervasive. La ridotta attività MTHFR compromette la sintesi di S-adenosilmetionina (SAM), il principale donatore di gruppi metile cellulare, necessario per la metilazione del DNA, delle proteine (inclusa la proteina basica della mielina) e dei lipidi. La carenza di SAM compromette inoltre la sintesi di glutatione, il principale antiossidante intracellulare e cofattore essenziale per gli enzimi di detossificazione dei metalli. Il risultato è una ridotta capacità di neutralizzare lo stress ossidativo indotto dall’alluminio e una compromessa detossificazione del metallo.
Le varianti dei geni della Glutatione S-Transferasi (GST), in particolare le delezioni nulle di GSTM1 e GSTT1 e il polimorfismo Ile105Val di GSTP1, rappresentano un terzo fattore critico. Questi enzimi sono fondamentali per la coniugazione del glutatione con composti elettrofili, inclusi i metalli pesanti, facilitando la loro eliminazione. Studi caso-controllo hanno dimostrato che le varianti GST sono associate a un triplice aumento del rischio di ASD e correlano direttamente con livelli ematici di alluminio più elevati nei soggetti affetti. Questi dati suggeriscono che l’incapacità di eliminare efficientemente l’alluminio, piuttosto che una maggiore esposizione, possa essere il fattore determinante in alcuni sottogruppi di pazienti.
Le varianti del sistema del complemento, in particolare gli alleli nulli di C4B, alterano le soglie della potatura sinaptica. Come discusso precedentemente, la carenza di C4B compromette l’attivazione efficiente del complemento classico, riducendo la capacità di marcare le sinapsi deboli per l’eliminazione microgliale. Questo si traduce in una sotto-potatura sinaptica con conseguente eccesso di spine dendritiche immature e circuiti corticali disfunzionali.
La convergenza di questi fattori genetici crea quella che può essere descritta come una “tempesta perfetta”: l’incapacità di detossificare i metalli si unisce a una risposta immunitaria iper-reattiva e a una disregolazione della potatura sinaptica, creando le condizioni biologiche per cui l’esposizione agli adiuvanti di alluminio si traduce in danno neurologico permanente.
La Sindrome ASIA e il Mimetismo Molecolare: Quando il Sistema Immunitario Sbaglia Bersaglio
La Sindrome Autoimmune Indotta da Adiuvanti (ASIA, Autoimmune/Inflammatory Syndrome Induced by Adjuvants), descritta per la prima volta da Shoenfeld e colleghi nel 2011, rappresenta un quadro clinico che raggruppa condizioni autoimmuni e neuroinfiammatorie scatenate dall’esposizione a adiuvanti contenuti in vaccini o altri prodotti. Questa sindrome offre un modello validato di patologia indotta dagli adiuvanti che presenta parallelismi striking con l’ipotesi vaccino-autismo.
I criteri diagnostici per la sindrome ASIA includono l’esposizione iniziale a un adiuvante (vaccino, impianto siliconico o altro prodotto contenente adiuvanti), una relazione temporale appropriata (tipicamente settimane o mesi post-esposizione), manifestazioni cliniche che includono malattie autoimmuni similari, sintomi neurologici o entrambe le categorie, e sierologia autoimmune positiva (autoanticorpi verso antigeni nucleari, antigeni tessuto-specifici o entrambi).
Le manifestazioni cliniche documentate nella sindrome ASIA comprendono sintomi neurologici quali mialgia, fatigue cronica, disfunzione cognitiva (“brain fog”), disturbi dell’umore e compromissione della memoria, oltre a manifestazioni autoimmuni che includono lupus eritematoso sistemico-simile, artrite reumatoide-simile, sindrome di Sjögren primaria e esacerbazione della miastenia grave. I marker infiammatori tipicamente elevati includono IL-1β, IL-6, TNF-α e indicatori di attivazione del complemento.
I parallelismi tra la sindrome ASIA e l’autismo sono impressionanti. Le manifestazioni neurologiche nell’ASD includono frequentemente fatigue cronica, disfunzione cognitiva (“brain fog”), compromissione della memoria e disturbi dell’umore quali ansia e depressione, tutti sintomi descritti nella sindrome ASIA. I processi simil-autoimmuni sono evidenti nell’ASD attraverso la presenza di autoanticorpi anti-cerebrali, anti-neuronali e anti-proteina basica della mielina in sottogruppi di pazienti. I marker infiammatori mostrano un overlap significativo, con elevate IL-1β, IL-6, TNF-α e evidenze di attivazione della via del complemento (alterazioni di C1q, C3, C4) sia nell’ASD che nella sindrome ASIA.
Il mimetismo molecolare rappresenta il meccanismo attraverso cui la risposta immunitaria diretta verso antigeni vaccinali può erroneamente dirigersi contro proteine self espresse nel sistema nervoso centrale. Questo fenomeno si verifica quando epitopi (brevi sequenze peptidiche riconosciute dai linfociti T e B) derivati da patogeni o vaccini presentano omologia strutturale con antigeni self, inducendo una risposta immune cross-reattiva che attacca i tessuti dell’ospite.
Diversi antigeni vaccinali dimostrano omologia di sequenza con proteine cerebrali. Gli epitopi della proteina del virus del morbillo condividono omologia con la proteina basica della mielina (MBP), un componente critico della guaina mielinica. Le risposte immunitarie specifiche per il morbillo indotte dal vaccino possono cross-reagire con la MBP, potenzialmente innescando demielinizzazione autoimmune. La tossina pertussis (componente del vaccino DTaP) è stata associata a encefalomielite post-vaccinale, con il mimetismo molecolare proposto come meccanismo attraverso cui la tossina o componenti batteriche correlate cross-reagiscono con antigeni neuronali nel sistema nervoso centrale. L’antigene di superficie dell’epatite B (HBsAg) dimostra omologia con la glicoproteina oligodendrocitaria della mielina (MOG), un componente critico della mielina del sistema nervoso centrale. Le risposte immunitarie all’epatite B possono cross-reagire con la MOG, innescando demielinizzazione autoimmune.
L’amplificazione di queste risposte cross-reattive da parte degli adiuvanti di alluminio rappresenta un elemento cruciale. L’attivazione dell’inflammasoma NLRP3 e la conseguente produzione di IL-1β promuovono la polarizzazione dei linfociti CD4+ naive verso il fenotipo Th17. I linfociti Th17 producono IL-17, IL-22 e altre citochine pro-infiammatorie, e mostrano una ridotta differenziazione verso il fenotipo regolatorio (Treg), manifestando un’autoreattività aumentata. L’IL-17 danneggia direttamente l’integrità della barriera emato-encefalica e facilita l’infiltrazione di cellule autoreattive nel sistema nervoso centrale. La ridotta tolleranza immunitaria mediata dalle cellule Treg rimuove il “freno” sulle risposte autoimmuni, permettendo ai cloni T autoreattivi generati durante l’epitope spreading di persistere e causare danno tissutale.
L’Epitope Spreading: Quando l’Attacco si Espande
L’epitope spreading rappresenta un meccanismo patogenetico attraverso il quale una risposta immunitaria inizialmente diretta verso un singolo antigene patogeno si espande progressivamente per includere epitopi su antigeni self correlati. Questo fenomeno spiega come la vaccinazione diretta verso un patogene specifico possa innescare risposte autoimmuni contro antigeni cerebrali che non condividono necessariamente omologia di sequenza diretta con l’antigene vaccinale.
Il processo di epitope spreading opera attraverso due meccanismi distinti: lo spreading intramolecolare, in cui la risposta si espande verso epitopi diversi sullo stesso antigene, e lo spreading intermolecolare, in cui la risposta si espande verso epitopi su antigeni distinti. Nel contesto della neuroinfammazione indotta dagli adiuvanti, lo spreading intermolecolare assume particolare rilevanza poiché può coinvolgere antigeni self espressi nel sistema nervoso centrale.
Il meccanismo proposto prevede che il rilascio di antigeni cerebrali durante il danno infiammatorio iniziale (causato dall’infiltrazione di cellule immunitarie e dall’eccitotossicità) fornisca nuovi target per il sistema immunitario. I frammenti di proteine cerebrali rilasciate vengono raccolte dalle cellule presentanti l’antigene e processate per la presentazione ai linfociti T. Se queste proteine condivido somiglianze strutturali con le proteine vaccinali inizialmente riconosciute, i cloni T cross-reattivi vengono attivati e proliferano. Il risultato è una risposta autoimmune che si espande progressivamente verso nuovi antigeni cerebrali, amplificando il danno iniziale e rendendo la patologia autosufficiente anche dopo la clearance dell’antigene vaccinale originale.
Questo meccanismo è particolarmente rilevante nel contesto dell’autismo, dove la regressione clinica osservata in molti casi potrebbe riflettere l’espansione progressiva della risposta autoimmune verso nuovi target cerebrali nel periodo successivo alla vaccinazione. La tempistica della regressione, tipicamente tra i 15 e i 30 mesi, è coerente con il tempo necessario per l’instaurarsi dell’epitope spreading e per il conseguente danno cumulativo.
L’Analisi secondo i Criteri di Bradford Hill: Un Quadro Causale Integrato
Il passaggio dall’ipotesi correlativa a un quadro causale integrato richiede la valutazione sistematica secondo i criteri di Bradford Hill, lo standard d’oro dell’epidemiologia per stabilire la causalità. Sir Austin Bradford Hill articolò nel 1965 nove criteri per giudicare quando le associazioni statistiche dovrebbero essere interpretate come causali, e questi criteri sono stati successivamente adottati dai tribunali nella valutazione delle rivendicazioni di danni tossicologici.
Il primo criterio, la forza dell’associazione, è soddisfatto dall’incremento di 80 volte della prevalenza di ASD e dall’infiltrazione linfocitaria 13 volte superiore nei cervelli autistici rispetto ai controlli. La grandezza di questi effetti supera quella di molti fattori di rischio ambientale consolidati e suggerisce un legame causale potente piuttosto che una semplice associazione spuria.
Il secondo criterio, la consistenza, è supportato dalla replicazione delle evidenze in molteplici specie (murini, bovini, umani), in studi condotti da gruppi di ricerca indipendenti in diversi paesi, e utilizzando metodologie diverse (studi in vitro, modelli animali, studi post-mortem, analisi epidemiologiche). L’attivazione dell’inflammasoma NLRP3 da parte dell’alluminio è stata confermata ripetutamente, così come l’elevazione delle citochine pro-infiammatorie nell’ASD.
Il terzo criterio, la specificità, è parzialmente soddisfatto dalla firma immunitaria distintiva osservata nei cervelli ASD: predominanza di linfociti CD8+ rispetto ai CD4+, espressione di granzima B, e localizzazione dell’infiltrazione nelle regioni cerebrali note per essere coinvolte nell’autismo (cortecce frontali, temporali e cervelletto). Tuttavia, la specificità assoluta è limitata poiché altri fattori possono teoricamente indurre meccanismi simili.
Il quarto criterio, la temporalità, è chiaramente soddisfatto. L’esposizione al carico cumulativo di alluminio picca tra la nascita e i 24 mesi, mentre le diagnosi di ASD si concentrano tra i 14 e i 36 mesi. I dati di sorveglianza indicano che la maggior parte degli eventi avversi neurologici gravi si verifica entro sei settimane dalla vaccinazione, coerente con la sequenza biologica attivazione dell’inflammasoma, compromissione della barriera emato-encefalica, infiltrazione immune, e danno neurologico.
Il quinto criterio, il gradiente biologico (dose-risposta), è supportato da evidenze su molteplici livelli. Modelli in vitro dimostrano che dosi crescenti di alluminio producono attivazione dell’inflammasoma proporzionalmente maggiore. Studi ecologici mostrano correlazioni positive tra il numero di vaccini contenenti alluminio nei calendari nazionali e la prevalenza di ASD attraverso tempo e geografia. Studi genetici mostrano che la “dose” di alleli di rischio (omozigosi HLA-DR4 rispetto a eterozigosi, omozigosi MTHFR 677TT rispetto a eterozigosi) correla con il rischio aumentato.
Il sesto criterio, la plausibilità, è soddisfatto dal meccanismo biologico dettagliatamente caratterizzato: fagocitosi dell’alluminio, attivazione dell’NLRP3, produzione di IL-1β, compromissione della barriera emato-encefalica, traslocazione macrofagica, attivazione microgliale, infiltrazione linfocitaria, attacco astrocitario, disregolazione della potatura sinaptica ed eccitotossicità. Ogni passaggio di questa cascata è supportato da letteratura peer-reviewed.
Il settimo criterio, la coerenza, è soddisfatto dall’allineamento tra dati epidemiologici (correlazione temporale), modelli animali (deficit motori e neuroinfiammazione con dosi equivalenti di alluminio), reperti post-mortem (infiltrati linfocitari, astrogliosi), e dati sierologici/citochinici (pattern infiammatorio caratteristico). L’insieme di queste evidenze forma un quadro coerente piuttosto che una collezione di anomalie inspiegabili.
L’ottavo criterio, l’evidenza sperimentale, è supportato da modelli di attivazione immune materna (MIA) che producono fenotipi simil-ASD nella prole attraverso l’attivazione del sistema immunitario durante la gravidanza. L manipolazione della segnalazione di IL-6 o IL-17A può attenuare o prevenire questi fenotipi, dimostrando la causalità del pathway infiammatorio. Modelli animali di esposizione all’alluminio dimostrano deficit motori, neuroinfiammazione e alterazioni comportamentali con dosi comparabili a quelle umane.
Il nono criterio, l’analogia, è soddisfatto dai paralleli con altre condizioni tossicologiche e cliniche. La sindrome ASIA offre un modello umano validato di patologia indotta dagli adiuvanti con manifestazioni neurologiche sovrapponibili a quelle dell’autismo. La neurotossicità di altri metalli quali piombo e mercurio avviene attraverso meccanismi di neuroinfiammazione, stress ossidativo e disregolazione della potatura sinaptica analoghi a quelli proposti per l’alluminio.
L’applicazione rigorosa dei criteri di Bradford Hill trasforma l’evidenza epidemiologica in un quadro causale integrato che soddisfa tutti i parametri. Questo risultato ha implicazioni profonde sia per la comprensione scientifica dell’ASD che per le implicazioni medico-legali e regolatorie.
Il Confronto con gli Standard Tossicologici Moderni: Un Divario Inaccettabile
Il divario tra gli standard tossicologici applicati ai farmaci e alle sostanze chimiche rispetto a quelli applicati agli adiuvanti vaccinali rivela un’incoerenza normativa che merita una critica severa. Per qualsiasi altra sostanza chimica destinata all’esposizione umana, in particolare durante periodi critici dello sviluppo, gli enti regolatori richiederebbero una valutazione completa degli effetti neurotossicologici prima dell’approvazione.
Gli standard EPA/OECD per la neurotossicità evolutiva richiedono la valutazione dell’impatto su neurogenesi e mielinizzazione, studi cronici a dosi ripetute per valutare la persistenza sistemica, la determinazione dell’accumulo cerebrale per sostanze che mostrano persistenza sistemica, e la valutazione degli effetti sul sistema nervoso centrale per sostanze che attivano citochine pro-infiammatorie. Tutti questi requisiti sono disattesi per gli adiuvanti di alluminio nel contesto vaccinale.
La giustificazione regolatoria tradizionale, ovvero che l’alluminio è stato utilizzato per decenni senza problemi evidenti, si scontra con almeno tre assunzioni storiche che sono state refutate dalla ricerca contemporanea. L’assunzione di clearance renale rapida è stata smentita dalla dimostrazione della biopersistenza macrofagica e della traslocazione al cervello. L’assunzione di impermeabilità della barriera emato-encefalica è stata smentita dai meccanismi di attraversamento CCL2-dipendente. L’assunzione di assenza di meccanismi che collegano l’attivazione immune periferica ai disturbi neuroevolutivi è stata smentita dalla caratterizzazione dell’asse immune-cervello e dei pathway infiammatori cerebrali.
Questo gap normativo non è semplicemente una lacuna tecnica, ma rappresenta un fallimento sistemico che ha permesso l’esposizione di milioni di bambini a potenziali neurotossici senza una valutazione adeguata dei rischi. La protezione legale dei produttori attraverso il NCVIA e le sue interpretazioni giurisprudenziali ha eliminato gli incentivi economici per la ricerca sulla sicurezza, mentre le agenzie regolatorie operano sotto vincoli statutari che impediscono l’imposizione di standard più rigorosi.
Implicazioni Economiche e Sociali: Il Costo dell’Inazione
La dimensione economica della crisi dell’autismo aggiunge urgenza alla necessità di una rivalutazione scientifica e normativa. Le stime dei costi lifetime per il supporto a un individuo con ASD negli Stati Uniti superano i 2,4 milioni di dollari, con costi aggregati per la società americana proiettati a 461 miliardi di dollari annui entro il 2025. Alcune proiezioni a lungo termine stimano costi futuri superiori a 1 trilione di dollari annui.
Questi costi riflettono molteplici componenti: le spese sanitarie per diagnosi, terapie e gestione delle comorbidità (anomalie motori con prevalenza del 79%, epilessia con prevalenza del 20%, disturbi gastrointestinali fino al 70%, disturbi del sonno nel 50-80% dei casi, condizioni psichiatriche nell’81% dei pazienti); i costi educativi per l’istruzione specializzata, gli interventi comportamentali e i servizi di supporto individuale; i costi sociali derivanti dalla disoccupazione cronica, che colpisce circa il 70% degli adulti autistici; e i costi informali sostenuti dalle famiglie per l’assistenza continuativa.
L’assenza di test mandatori di neurotossicità evolutiva che potrebbero identificare sostanze interferenti con i processi di neurosviluppo rappresenta un costo occulto che si aggiunge a questi oneri diretti. Se l’alluminio adiuvante contribuisse anche solo parzialmente all’epidemia di ASD, l’investimento iniziale necessario per sviluppare adiuvanti alternativi più sicuri e per condurre studi epidemiologici adeguati risulterebbe trascurabile rispetto ai costi evitabili della disabilità permanente.
Prospettive e Raccomandazioni: Verso una Medicina Preventiva
Alla luce delle evidenze analizzate, emergono alcune direzioni prioritarie per la ricerca futura e la politica regolatoria. La ricerca dovrebbe concentrarsi sullo sviluppo e la validazione di adiuvanti alternativi non neurotossici, sull’identificazione di biomarcatori di suscettibilità genetica che permettano una medicina personalizzata, su studi prospettici di coorte che esaminino gli esiti neuroevolutivi in funzione del carico cumulativo di alluminio, e sulla caratterizzazione dei meccanismi di reversibilità o prevenzione del danno.
Dal punto di vista regolatorio, le raccomandazioni includono l’obbligatorietà di studi di neurotossicità evolutiva per tutti gli adiuvanti nuovi ed esistenti, con monitoraggio della biopersistenza cerebrale per almeno 12 mesi. Sarebbe opportuno integrare lo screening dei polimorfismi MTHFR, HLA-DR4, GST e varianti C4B nelle fasi di approvazione per identificare sottogruppi di popolazione a rischio sproporzionato. È necessaria una ricalibrazione dei limiti di alluminio per dose (attualmente 0,85 mg), definendo nuovi limiti basati sulla sicurezza del neurosviluppo parametrati specificamente sulla finestra 0-18 mesi. Infine, è opportuno riconsiderare il principio di “grandfathering” alla luce delle nuove evidenze scientifiche, applicando standard tossicologici moderni anche agli adiuvanti storici.
Dal punto di vista clinico, la consapevolezza dei fattori di rischio genetici potrebbe informare discussioni più personalizzate sulla tempistica e la composizione dei calendari vaccinali per bambini con vulnerabilità identificate. L’identificazione precoce di segni di neuroinfiammazione potrebbe permettere interventi tempestivi per mitigare il danno in corso. L’integrazione di strategie di supporto metabolico (ad esempio, supplementazione con metilfolato e glutatione) potrebbe ridurre la vulnerabilità in portatori di polimorfismi MTHFR.
Conclusione: L’Imperativo dell’Azione
La convergenza di evidenze epidemiologiche, meccanicistiche, neuropatologiche e genetiche delinea un quadro che non può più essere ignorato. L’incremento di 80 volte della prevalenza di ASD, la correlazione temporale quasi perfetta con l’espansione del calendario vaccinale, i meccanismi biologici dettagliatamente caratterizzati di traslocazione dell’alluminio al cervello e attivazione dell’inflammasoma NLRP3, le evidenze neuropatologiche di attacco immunitario attivo ai tessuti cerebrali, e la vulnerabilità genetica identificata in una significativa proporzione di casi formano un corpus di conoscenze che impone una risposta.
La persistenza di un quadro normativo basato su assunzioni scientificamente obsolete, combinata con la protezione legale dei produttori che elimina gli incentivi alla sicurezza, rappresenta un fallimento sistemico che ha permesso l’esposizione di una generazione intera a potenziali neurotossici senza adeguata valutazione del rischio. La responsabilità istituzionale impone un cambiamento di paradigma che ponga la sicurezza neurologica al di sopra della semplificazione procedurale.
Non si tratta di demonizzare la vaccinazione, una delle più importanti conquiste della sanità pubblica che ha salvato innumerevoli vite da malattie infettive devastanti. Si tratta piuttosto di riconoscere che la composizione dei vaccini, in particolare gli adiuvanti, merita la stessa attenzione rigorosa alla sicurezza che richiediamo per qualsiasi altra sostanza destinata all’esposizione umana, specialmente durante le finestre critiche dello sviluppo neurologico. La salute dei nostri bambini merita nulla di meno.
La scienza non è mai definitiva, ma è obbligata a seguire le evidenze dove esse conducono. Nel caso degli adiuvanti di alluminio e dell’autismo, le evidenze conducono verso una rivalutazione urgente e una maggiore umiltà epistemica su ciò che non sappiamo. L’inazione, di fronte a prove crescenti di danno potenziale, non è neutralità scientifica ma complicità passiva in un sistema che ha sistematicamente privilegiato la convenienza sulla sicurezza.
Riferimenti Bibliografici
1.Mechanisms of NLRP3 inflammation activation by aluminum adjuvants. Frontiers in Immunology, 15:1234567. https://www.frontiersin.org/articles/10.3389/fimmu.2025.1234567
2.Araszkiewicz, P., et al. (2025). MTHFR polymorphisms and autism spectrum disorder: A systematic review. Molecular Genetics and Metabolism, 134(2): 145-158. https://www.sciencedirect.com/science/article/pii/S1096715925001234
3.Ashwood, P., et al. (2011). The role of immune dysfunction in the pathophysiology of autism. Brain, Behavior, and Immunity, 25(3): 395-399. https://www.sciencedirect.com/science/article/pii/S0889159111000067
4.Baylor, N.W., et al. (2002). Aluminum salts in vaccines—US perspective. Vaccine, 20(Suppl 3): S18-S23. https://www.sciencedirect.com/science/article/pii/S0264410X02000788
5.Blaylock, R.L. (2015). Aluminum induced immunoexcitotoxicity in autism spectrum disorder. Journal of Neurological Disorders, 3(4): 215. https://www.jneurologicaldisorders.com/articles/aluminum-induced-immunoexcitotoxicity-autism-spectrum-disorder
6.Boris, M., et al. (2004). Association of MTHFR gene variants with autism. Journal of American Physicians and Surgeons, 9(4): 106-108. https://japsonline.com/archives/2004/oct/japs-2004-04-02.pdf
7.Braunschweig, D., et al. (2013). Autism: Maternally derived antibodies specific for fetal brain proteins. Neurotoxicology, 35: 161-168. https://www.sciencedirect.com/science/article/pii/S0161813X13000489
8.Brimberg, L., et al. (2016). Antibodies as mediators of brain pathology. Trends in Immunology, 37(11): 758-770. https://www.sciencedirect.com/science/article/pii/S1471490616300792
9.Brüsewitz v. Wyeth LLC, 562 U.S. 223 (2011). https://supreme.justia.com/cases/federal/us/562/223/
10.CDC (2025). Autism Spectrum Disorder Surveillance — 2025. https://www.cdc.gov/ncbddd/autism/data.html
11.Choi, G.B., et al. (2016). The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science, 351(6276): 933-939. https://science.sciencemag.org/content/351/6276/933
12.Crépeaux, G., et al. (2017). Non-linear dose-response of aluminium hydroxide adjuvant particles: A literature review. Current Medicinal Chemistry, 24(7): 611-622. https://www.ingentaconnect.com/content/ben/cmc/2017/00000024/00000007
13.Deoni, S.C., et al. (2016). Mapping infant brain myelination with MRI. NeuroImage, 141: 133-142. https://www.sciencedirect.com/science/article/pii/S1053811916302313
14.DiStasio, M.M., et al. (2019). CD8+ T cells and CD8+/CD4+ T cell ratios in autism: Postmortem brain evidence. Journal of Neuroinflammation, 16(1): 70. https://jneuroinflammation.biomedcentral.com/articles/10.1186/s12974-019-1472-x
15.Edmonson, C., et al. (2014). Elevated GFAP expression in brains of individuals with autism spectrum disorder. Molecular Autism, 5(1): 51. https://molecularautism.biomedcentral.com/articles/10.1186/2040-2392-5-51
16.Eisenbarth, S.C., et al. (2008). Crucial role for the Nalp3 inflammasome in the adjuvant effects of aluminium hydroxide. Nature Immunology, 9(8): 847-848. https://www.nature.com/articles/ni.1631
17.Ellul, P., et al. (2021). Th17 and Treg imbalance in autism spectrum disorder. Cytokine, 138: 155327. https://www.sciencedirect.com/science/article/pii/S1043466620302697
18.Flarend, R.E., et al. (1997). In vivo absorption of aluminium-containing vaccine adjuvants using 26Al. Vaccine, 15(12-13): 1314-1318. https://www.sciencedirect.com/science/article/pii/S0264410X97000862
19.Frye, R.E., et al. (2013). Folate receptor alpha autoantibodies in autism spectrum disorder. Molecular Autism, 4(1): 26. https://molecularautism.biomedcentral.com/articles/10.1186/2040-2392-4-26
20.Ganz, M.L. (2007). The lifetime distribution of the incremental societal costs of autism. Archives of Pediatrics & Adolescent Medicine, 161(4): 343-349. https://jamanetwork.com/journals/jamapediatrics/fullarticle/209172
21.Geier, D.A., et al. (2004). Thimerosal in childhood vaccines, neurodevelopment disorders, and heart disease in the United States. Journal of Toxicology and Environmental Health, 66(8): 677-686. https://www.tandfonline.com/doi/abs/10.1080/15287390490428609
22.Gherardi, R.K., et al. (2015). Biopersistence and brain translocation of aluminium in vaccine-injected mice. Current Medicinal Chemistry, 22(34): 3934-3938. https://www.ingentaconnect.com/content/ben/cmc/2015/00000022/00000034
23.Goldman, G.S., et al. (2025). Polysorbate 80 in vaccines: Pharmacokinetics and safety considerations. Vaccine, 43(1): 123-135. https://www.sciencedirect.com/science/article/pii/S0264410X25000123
24.Grotheer, M., et al. (2022). Myelin development in human infancy and early childhood. NeuroImage, 250: 118900. https://www.sciencedirect.com/science/article/pii/S1053811922000753
25.Gunaydin, H., et al. (2025). Complement system dysregulation in autism spectrum disorder. Frontiers in Immunology, 16: 876543. https://www.frontiersin.org/articles/10.3389/fimmu.2025.876543
26.Hassan, A.M., et al. (2025). Autoantibodies against myelin proteins in autism: A systematic review. Autoimmunity Reviews, 24(3): 103456. https://www.sciencedirect.com/science/article/pii/S1568997225000345
27.Hill, A.B. (1965). The environment and disease: Association or causation? Proceedings of the Royal Society of Medicine, 58(5): 295-300. https://journals.sagepub.com/doi/10.1177/003591576505800502
28.Hooker, B., et al. (2020). Positive association between aluminum vaccine adjuvants and neurodevelopmental disorders. International Journal of Environmental Research and Public Health, 17(18): 6879. https://www.mdpi.com/1660-4601/17/18/6879
29.Hoxhaj, G., et al. (2022). Vaccine schedules and autism: A population-based study. Journal of Public Health, 44(2): 456-465. https://academic.oup.com/jpubhealth/article/44/2/456/6530121
30.Huttenlocher, P.R., et al. (1990). Synaptogenesis, reduction in relation to brain growth. Proceedings of the National Academy of Sciences, 87(16): 6146-6150. https://www.pnas.org/content/87/16/6146
31.IOM (Institute of Medicine). (2012). Adverse Effects of Vaccines: Evidence and Causality. Washington, DC: The National Academies Press. https://www.nap.edu/catalog/13195/adverse-effects-of-vaccines-evidence-and-causality
32.James, S.J., et al. (2006). Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. American Journal of Clinical Nutrition, 80(6): 1611-1617. https://academic.oup.com/ajcn/article/80/6/1611/469034
33.Janeway, C.A., et al. (2001). Immunobiology (5th ed.). Garland Science. https://www.garlandscience.com/textbook/9780815341016
34.Jones, K., et al. (2024). Acetaminophen use in infancy and autism risk: A review of the evidence. Neurotoxicology, 105: 123-134. https://www.sciencedirect.com/science/article/pii/S0161813X24000123
35.Kebir, H., et al. (2007). Endothelin-1 regulates brain endothelial cell permeability through IL-17A dependent pathway. Nature Medicine, 13(10): 1173-1175. https://www.nature.com/articles/nm1638
36.Khan, Z., et al. (2013). Slow CCL2-dependent translocation of biopersistent particles from muscle to brain. BMC Medicine, 11: 99. https://bmcmedicine.biomedcentral.com/articles/10.1186/1741-7015-11-99
37.Kivity, S., et al. (2009). Infections and vaccines in the etiology of central nervous system demyelinating diseases. Autoimmunity Reviews, 8(4): 332-336. https://www.sciencedirect.com/science/article/pii/S1568997208002546
38.Korn, T., et al. (2007). IL-17 and Th17 cells. Annual Review of Immunology, 27: 485-517. https://www.annualreviews.org/doi/10.1146/annurev.immunol.021908.132710
39.Lau, A., et al. (2010). Glutamate receptors, neurotoxicity and neurodegeneration. Pflügers Archiv – European Journal of Physiology, 460(2): 525-542. https://link.springer.com/article/10.1007/s00424-010-0809-1
40.Li, H., et al. (2008). Aluminum salts induce the maturation of dendritic cells in vitro. Cellular Immunology, 251(2): 81-91. https://www.sciencedirect.com/science/article/pii/S0008876308001792
41.Li, X., et al. (2009). Elevated immune response in the brain of autistic patients. Journal of Neuroimmunology, 207(1-2): 111-116. https://www.sciencedirect.com/science/article/pii/S0165572808003494
42.Liao, X., et al. (2020). Cytokine alterations in autism spectrum disorder: A systematic review and meta-analysis. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 98: 109775. https://www.sciencedirect.com/science/article/pii/S0278584619303945
43.Lyons-Weiler, J., et al. (2022). Cumulative aluminum exposure from vaccines and synaptic pruning: A mechanistic hypothesis. Journal of Trace Elements in Medicine and Biology, 71: 126942. https://www.sciencedirect.com/science/article/pii/S0946672X22000423
44.Marrack, P., et al. (2009). Towards an understanding of the adjuvant action of aluminium. Nature Reviews Immunology, 9(4): 287-293. https://www.nature.com/articles/nri2510
45.Masson, J.D., et al. (2022). Neuroinflammation induced by aluminum adjuvants: A systematic review. Neurotoxicology, 90: 34-47. https://www.sciencedirect.com/science/article/pii/S0161813X22000526
46.Mawson, A.R., et al. (2017). Pilot comparative study on the health of vaccinated and unvaccinated 6- to 12-year-old U.S. children. Journal of Translational Science, 3(3): 1-12. https://www.jtranslationalmedicine.com/articles/10.14205/2305-5820.03.03.161
47.Meltzer, A., et al. (2017). Cytokine dysregulation in autism spectrum disorder. Neuroscience & Biobehavioral Reviews, 82: 729-749. https://www.sciencedirect.com/science/article/pii/S0149763417304171
48.Miller, N.Z. (2016). Aluminum in childhood vaccines: Exposure and cumulative dose. Advances in Experimental Medicine and Biology, 848: 123-139. https://link.springer.com/chapter/10.1007/978-3-319-21314-2_7
49.Moaaz, M., et al. (2019). Th17/Treg imbalance in autism spectrum disorder. Journal of Neuroimmunology, 337: 577067. https://www.sciencedirect.com/science/article/pii/S0165572819303775
50.Modabbernia, A., et al. (2017). Environmental risk factors for autism: An evidence-based review of systematic reviews and meta-analyses. Molecular Autism, 8: 13. https://molecularautism.biomedcentral.com/articles/10.1186/s13229-017-0121-4
51.Morgan, J.T., et al. (2010). Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biological Psychiatry, 68(4): 368-376. https://www.sciencedirect.com/science/article/pii/S0006322310004175
52.Mostafa, G.A., et al. (2010). The link between autoimmunity and autism: A new review. Autoimmunity Reviews, 9(11): 728-732. https://www.sciencedirect.com/science/article/pii/S1568997210001578
53.Nevison, C.D. (2014). A comparison of temporal trends in United States autism prevalence to trends in suspected environmental factors. Environmental Health, 13: 73. https://ehjournal.biomedcentral.com/articles/10.1186/1476-069X-13-73
54.Nirenberg, M.S. (2025). Vaccine adjuvants and autism: An update on the depot hypothesis. Vaccine, 43(2): 234-245. https://www.sciencedirect.com/science/article/pii/S0264410X25000189
55.Odell, J.D., et al. (2005). Correlation of C4B null alleles with autism. Clinical Immunology, 116(3): 249-252. https://www.sciencedirect.com/science/article/pii/S1521661605002299
56.Parker, W., et al. (2017). Acetaminophen, vaccines, and autism: Evidence for a behavioral phenotype. Journal of Trace Elements in Medicine and Biology, 42: 29-33. https://www.sciencedirect.com/science/article/pii/S0946672X17300308
57.Parker, W., et al. (2023). Acetaminophen and autism: An updated hypothesis. Journal of Trace Elements in Medicine and Biology, 80: 127234. https://www.sciencedirect.com/science/article/pii/S0946672X23000987
58.Perricone, C., et al. (2013). ASIA syndrome and autoimmune/inflammatory syndrome induced by adjuvants: Overlap and difference. Clinical Reviews in Allergy & Immunology, 47(2): 241-247. https://link.springer.com/article/10.1007/s12016-013-8402-y
59.Petrovsky, N., et al. (2015). Vaccine adjuvant safety: The elephant in the room. Expert Review of Vaccines, 14(2): 147-149. https://www.tandfonline.com/doi/full/10.1586/14760584.2015.986463
60.Presumey, J., et al. (2017). Complement system in neural synapse elimination in development and disease. Advances in Immunology, 135: 53-79. https://www.sciencedirect.com/science/article/pii/S0065277617300159
61.Rahbar, M.H., et al. (2015). Interaction between glutathione S-transferase variants and blood lead levels in autism. Journal of Autoimmunity, 62: 56-62. https://www.sciencedirect.com/science/article/pii/S0896841115000509
62.Rossignol, D.A., et al. (2014). Environmental toxicants and autism spectrum disorders: A systematic review. Translational Psychiatry, 4(2): e360. https://www.nature.com/articles/tp2013132
63.Saghazadeh, A., et al. (2019). A meta-analysis of pro-inflammatory cytokines in autism spectrum disorders: Effects of age, gender, and latitude. Journal of Psychiatric Research, 115: 90-102. https://www.sciencedirect.com/science/article/pii/S0022395618308199
64.Sandin, S., et al. (2014). The familial risk of autism. JAMA, 311(17): 1770-1777. https://jamanetwork.com/journals/jama/fullarticle/1866699
65.Schafer, D.P., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron, 74(4): 691-705. https://www.sciencedirect.com/science/article/pii/S0896627312004641
66.Sejvar, J.J., et al. (2003). Neurologic adverse events associated with vaccination. Current Neurology and Neuroscience Reports, 3(3): 195-203. https://link.springer.com/article/10.1007/s11910-003-0078-3
67.Semple, B.D., et al. (2010). Elevated IL-1β and IL-6 in autism spectrum disorders. Journal of Neuroimmunology, 229(1-2): 198-201. https://www.sciencedirect.com/science/article/pii/S0165572809003894
68.Shaw, C.A., et al. (2013). Aluminum-induced entropy in biological systems: Implications for neurological disease. Journal of Toxicology, 2013: 491316. https://www.hindawi.com/journals/jt/2013/491316/
69.Shoenfeld, Y., et al. (2011). ‘ASIA’ – Autoimmune/inflammatory syndrome induced by adjuvants. Journal of Autoimmunity, 36(1): 4-8. https://www.sciencedirect.com/science/article/pii/S0896841110001755
70.Smith, S.E., et al. (2007). Maternal immune activation alters offspring behavior. Brain, Behavior, and Immunity, 21(6): 715-725. https://www.sciencedirect.com/science/article/pii/S0889159106002385
71.Stephan, A.H., et al. (2012). A prominent role for astrocyte-mediated complement in synaptic pruning. Annual Review of Neuroscience, 35: 411-432. https://www.annualreviews.org/doi/10.1146/annurev-neuro-062012-170330
72.Tang, G., et al. (2014). Synaptic pruning in the female mutant mice. Nature, 506(7487): 110-114. https://www.nature.com/articles/nature12975
73.Torres, A.R., et al. (2002). HLA-DR4 in families with autism. Journal of Autism and Developmental Disorders, 32(3): 173-178. https://link.springer.com/article/10.1023/A:1015432206709
74.Torres, A.R., et al. (2016). The association of HLA alleles in autism. Human Immunology, 77(10): 902-907. https://www.sciencedirect.com/science/article/pii/S0198885916301781
75.Vallese, C., et al. (2024). NLRP3 inflammasome activation and mitochondrial dysfunction in autism spectrum disorder. Cell Death & Disease, 15(4): 234. https://www.nature.com/articles/s41419-024-06789-5
76.Vargas, D.L., et al. (2005). Neuroglial activation and neuroinflammation in the brain of patients with autism. Annals of Neurology, 57(1): 67-81. https://onlinelibrary.wiley.com/doi/10.1002/ana.20315
77.Wang, J., et al. (2025). Polysorbate 80-mediated blood-brain barrier modulation for drug delivery. Advanced Drug Delivery Reviews, 197: 114801. https://www.sciencedirect.com/science/article/pii/S0169409X25000123
78.Warren, R.P., et al. (1994). C4 null alleles in autism. Clinical Immunology and Immunopathology, 72(2): 238-241. https://www.sciencedirect.com/science/article/pii/0090122994902299
79.Wei, H., et al. (2011). Cytokine expression in autism spectrum disorder. Neurochemical Research, 36(10): 1964-1970. https://link.springer.com/article/10.1007/s11064-011-0544-5
80.Xiong, W., et al. (2023). Complement-mediated synaptic pruning in neurodevelopment and neuropsychiatric disorders. Neuroscience Bulletin, 39(3): 435-452. https://link.springer.com/article/10.1007/s12264-022-00966-y

Lasciate il vostro commento